


Como é organizada a documentação da gestão do risco?
A documentação desenvolvida no âmbito do sistema da gestão de risco é organizada num arquivo da gestão de risco. Este arquivo deve conter um plano de gestão do risco para cada dispositivo, uma matriz da gestão do risco e um relatório da gestão do risco.
O plano da gestão do risco é um elemento essencial no sistema de gestão do risco e descreve:
- O processo de gestão do risco ao longo do ciclo de vida do dispositivo
- As responsabilidades e autoridades do pessoal
- Os requisitos para revisão das atividades de gestão do risco
- A política da gestão do risco
- O método de avaliação do risco residual global
- A verificação da implementação das medidas de controlo do risco
- A identificação das fontes de informação de produção e pós-produção
É essencial estabelecer uma política de gestão do risco uma vez que proporciona um quadro para a definição de critérios de aceitabilidade do risco, ao determinar níveis de probabilidade e severidade que serão utilizados para estimar riscos na matriz do risco. A definição e documentação da política de gestão do risco é da responsabilidade da gestão de topo.
A matriz da gestão do risco encontra-se associada ao respetivo plano e relatório, e funciona como folha de trabalho para o processo de gestão do risco, permitindo a rastreabilidade dos riscos.
Sempre que surgem novas informações, quando a conceção, desenvolvimento e fabrico do dispositivo são alterados, ou sempre que o responsável pela observância da regulamentação (PRRC na sigla em inglês) considerar necessário, é elaborado um relatório da gestão do risco que inclui os resultados da revisão das atividades de gestão do risco. O relatório da gestão do risco compreende:
- Os resultados da avaliação do risco
- As medidas de controlo do risco implementadas
- A aceitabilidade dos riscos e a análise benefício/risco para os riscos não aceitáveis
- A avaliação da aceitabilidade do risco residual global e medidas de controlo adicionais
- A revisão da informação de produção e pós-produção
Como é desenvolvido e mantido o processo de gestão do risco?
A Norma ISO 14971:2019, que substitui a norma EN ISO 14971:2012, apresenta um quadro para desenvolver e manter um processo de gestão do risco de forma sistemática. A Norma ISO 14971:2019 ainda não se encontra harmonizada com o RDM UE, no entanto, a Comissão Europeia já publicou um esboço do pedido de harmonização às organizações de Harmonização Europeia (CEN e CENELEC).
A aplicação da ISO 14971:2019 é orientada pelo relatório técnico ISO/TR 24971:2020. A ISO 14971:2019 correlaciona-se ainda com a Norma IEC 62366-1:2015, relativa à aplicação de engenharia de usabilidade a dispositivos médicos. De acordo com a ISO 14971:2019, o processo de gestão do risco inclui: análise do risco, avaliação do risco, controlo do risco, avaliação do risco residual global, revisão da gestão do risco e atividades de produção e pós-produção (Figura 1).
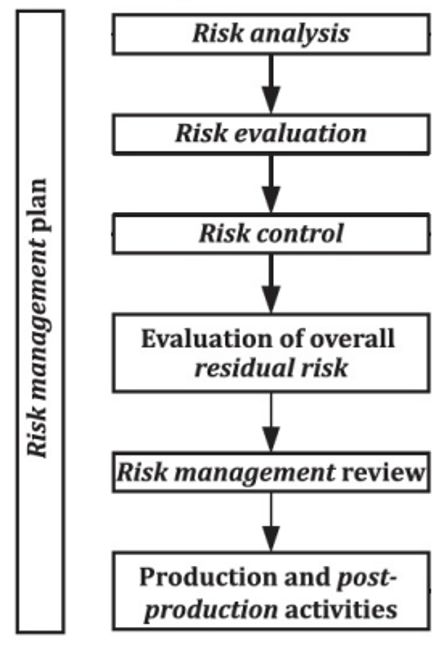
Figura 1. Passos do processo de gestão do risco, adaptado de ISO/TR 24971:2020
Análise do risco
O primeiro passo da análise do risco é documentar a utilização prevista e a má utilização razoavelmente previsível do dispositivo e identificar as características quantitativas e qualitativas relacionadas com a sua segurança. A utilização prevista considera a indicação médica, a população de doentes, a parte do corpo que interage com o dispositivo, o perfil do utilizador, o ambiente de utilização e o princípio de funcionamento do dispositivo. A má utilização razoavelmente previsível é uma nova característica presente na ISO 14971:2019, e é definida pela utilização do dispositivo médico de uma forma não prevista pelo Fabricante, mas que pode ser resultado de um comportamento humano previsível, como erros de utilização, atos intencionais de má utilização e utilização do dispositivo para outras finalidades médicas não previstas. Os casos de má utilização razoavelmente previsível podem ser determinados aplicando um processo de engenharia da usabilidade, de acordo com a IEC 62366-1:2015.
O Fabricante deve então analisar os perigos previsíveis e examinar as sequências de eventos que podem resultar numa situação perigosa. A probabilidade de ocorrer uma situação perigosa é calculada através do produto das probabilidades de ocorrência de cada evento independente (Figura 2).
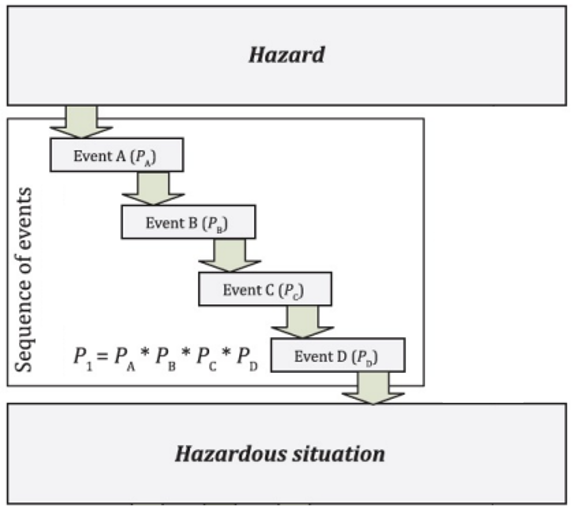
Figura 2. Relação entre perigos, sequência de eventos e situação perigosa, adaptado de ISO/TR 24971:2020
Finalmente, os riscos são estimados, atribuindo níveis de probabilidade e severidade a cada situação perigosa, de acordo com os critérios definidos na política de gestão do risco. Se existirem dados suficientes, a probabilidade deve ser expressa quantitativamente, caso contrário, é preferível aplicar um método qualitativo. Apesar da probabilidade ser uma variável contínua, pode ser decomposta em níveis discretos, sendo que quando não pode ser estimada, o risco é avaliado exclusivamente com base na severidade. A severidade de um dano é um continuum, mas também pode ser decomposta em níveis discretos. O Fabricante decide o número de níveis da probabilidade e da severidade, sendo que a matriz resultante é frequentemente 3×3 ou 5×5.
Avaliação do risco
O Fabricante avalia cada risco estimado e determina a sua aceitabilidade com base nos critérios definidos na política de gestão do risco, documentada no plano de gestão do risco. Caso o risco seja aceitável, não serão necessárias medidas de controlo do risco e o risco será tratado como residual.
Controlo do risco
Caso o risco seja inaceitável ou condicionalmente aceitável, deve ser mitigado. Existem duas abordagens ao controlo do risco.
O fabricante pode abordar o controlo de risco com base na praticabilidade das medidas de risco, estabelecendo ponderações entre aceitar certos riscos e a disponibilidade dos dispositivos no mercado.
Outra abordagem é baseada na magnitude do risco residual, na qual o Fabricante tenta reduzir os riscos tanto quanto possível, sem afetar adversamente a relação benefício/risco. Existem três opções para controlo do risco, que são implementadas por ordem prioritária. Primeiro, o Fabricante concebe e fabrica o dispositivo de forma inerentemente segura. Segundo, o Fabricante adota medidas protetivas no dispositivo ou no processo de fabrico. Terceiro, o Fabricante emite informação de segurança ou treino ao utilizador.
Após implementação das medidas de controlo do risco os riscos individuais são avaliados, utilizando novamente os critérios de aceitabilidade definidos na política de gestão do risco. Caso os riscos permaneçam inaceitáveis e nenhuma outra medida de controlo seja aplicável, é realizada uma análise benefício/risco baseada em dados e na literatura. Caso o benefício não supere o risco, o Fabricante deve considerar modificar o dispositivo ou a sua utilização prevista.
Algumas medidas de controlo podem introduzir novos riscos ou alterar riscos já identificados, por isso o Fabricante deve rever os efeitos das medidas de controlo do risco.
Avaliação do risco residual global
Após implementação e verificação das medidas de controlo do risco, o fabricante avalia o risco residual global, utilizando o método e os critérios definidos no plano de gestão do risco. O método para avaliar o risco residual global pode incluir: a ponderação entre os benefícios e o risco residual global, a representação visual dos riscos residuais, a comparação com dispositivos médicos semelhantes, uma avaliação de peritos e investigações adicionais.
Se o risco residual global for considerado inaceitável, o fabricante pode considerar a implementação de medidas adicionais de controlo do risco ou a modificação do dispositivo ou da utilização prevista.
Revisão da gestão do risco
Antes de libertar o dispositivo para comercialização, o Fabricante confirma que a gestão do risco foi devidamente executada e que os resultados se encontram registados no relatório da gestão do risco, que o risco residual global é considerado aceitável e que se encontram aplicados métodos para recolher e rever informação de produção e pós-produção relevante.
Atividades de produção e pós-produção
O Fabricante reúne e revê informação de produção e pós-produção, incluindo informação gerada durante a produção e durante a monitorização do processo de produção, informação gerada pelo utilizador, informação de instalação, utilização e manutenção do dispositivo, informação gerada pela cadeia de fornecimento, informação disponível publicamente e informação relacionada com o estado da arte.
Caso a informação recolhida seja determinante para a segurança, o Fabricante pode aplicar ações relativas ao dispositivo ou ao processo da gestão do risco. As ações podem ser direcionadas a dispositivos já distribuídos, dispositivos já fabricados mais ainda não distribuídos ou dispositivos que ainda não foram fabricados.
Como é que a Norma IEC 62366 se correlaciona com a ISO 14971:2019?
A secção 5 do capítulo 1 do Anexo I do RDM UE exige que o Fabricante reduza, tanto quanto possível, os riscos associados às características ergonómicas do dispositivo e ao meio em que este se destina a ser utilizado e que sejam considerados os conhecimentos técnicos, a experiência, a educação, a formação e o ambiente de utilização, e as condições clínicas e físicas dos utilizadores previstos.
Assim, a Norma IEC 62366-1:2015 auxilia o Fabricante na análise, especificação, desenvolvimento e avaliação da usabilidade relacionada com a segurança do dispositivo médico. O processo de engenharia da usabilidade permite que o Fabricante determine e mitigue os riscos associados à utilização normal do dispositivo, que inclui a utilização correta e erros de utilização. O processo pode ser também utilizado para identificar, mas não mitigar riscos associados à utilização anormal do dispositivo.
O objetivo do relatório técnico IEC 62366-2:2016 é mais geral, focando-se não apenas na usabilidade relacionada coma segurança, mas também na usabilidade relacionada com atributos como a precisão, completude e eficiência das tarefas, e satisfação do utilizador.
Referências:
- Regulamento (UE) 2017/745, sobre dispositivos médicos
- ISO 14971:2019, sobre a aplicação da gestão do risco a dispositivos médicos
- ISO/TR 24971:2020, orientação para aplicação da ISO 14971
- IEC 62366-1:2015, sobre a aplicação da engenharia de usabilidade a dispositivos médicos











