


How is risk management documentation organized?
The documentation developed in the scope of the risk management system is organized in a risk management file. This archive should include a risk management plan for each device, a risk management matrix and a risk management report.
The risk management plan is a key element of the risk management system and it describes:
- the risk management process throughout the lifecycle of the device
- the responsibilities and authorities of the personnel
- the requirements for revision of risk management activities
- the risk management policy
- the method for evaluating the overall residual risk
- the verification of implementation of risk control measures
- the identification of sources of production and post-production information
Establishing a risk management policy is of particular importance because it provides a framework for the definition of criteria for risk acceptability, by determining levels for risk probability and severity which will be used to estimate risk in the risk matrix. It is the responsibility of top management to define and document the risk management policy.
The risk management matrix is linked to the respective risk management plan and report, functioning as worksheet for the risk management process allowing risk traceability.
When new information emerges, in case of any change to the design, development and manufacture of the device or anytime the person responsible for regulatory compliance (PRRC) considers it necessary, a risk management report comprising the results from the review of risk management activities should be developed. The risk management report contains:
- the results of risk evaluation
- the risk control measures implemented
- the risk acceptability and benefit/risk analysis of the unacceptable risks
- the acceptability evaluation of overall residual risk and additional control measures
- the revision of production and post-production information
How is the risk management process developed and maintained?
A systematic framework to develop and maintain a risk management process is presented in standard ISO 14971:2019, which supersedes EN ISO 14971:2012. The standard ISO 14971:2019 is not yet harmonized under EU MDR, but the European Commission already published a draft of the standardization request to the European Standards organizations (CEN and CENELEC).
The application of ISO 14971:2019 is guided by the technical report ISO/TR 24971:2020. The ISO 14971:2019 is correlated with the standard IEC 62366-1:2015, regarding the application of usability engineering to medical devices.
According to ISO 14971:2019, the risk management process comprehends: risk analysis, risk evaluation, risk control, evaluation of overall residual risk, risk management review and production and post-production activities (Figure 1).
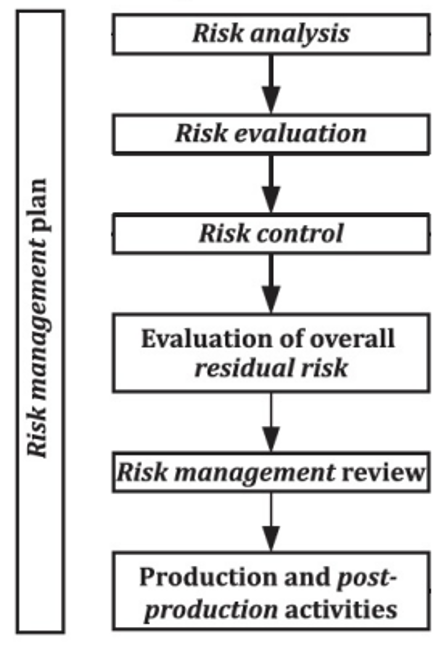
Figure 1. Risk management process steps, adapted from ISO/TR 24971:2020.
Risk analysis
The first step of risk analysis is to document the intended use and the reasonably foreseeable misuse of the device and identify the quantitative and qualitative characteristics related to its safety. The intended use takes into account the medical indication, the patient population, the part of the body interacting with the device, the user profile, the use environment and the operating principle of the device. The reasonably foreseeable misuse is a new feature presented in ISO 14971:2019, and it is defined as the use of the medical device in a way not intended by the Manufacturer, but which can result from predictable human behaviour, such as use errors, intentional acts of misuse, and use of the medical device for other non-intended medical applications. Cases of reasonably foreseeable misuse can be determined by applying a usability engineering process, according to IEC 62366-1:2015.
The Manufacturer shall then analyse the foreseeable hazards and examine sequences of events which can result in hazardous situations. The probability of occurrence of a hazardous situation is given by the product of probabilities of occurrence of each independent event (Figure 2).
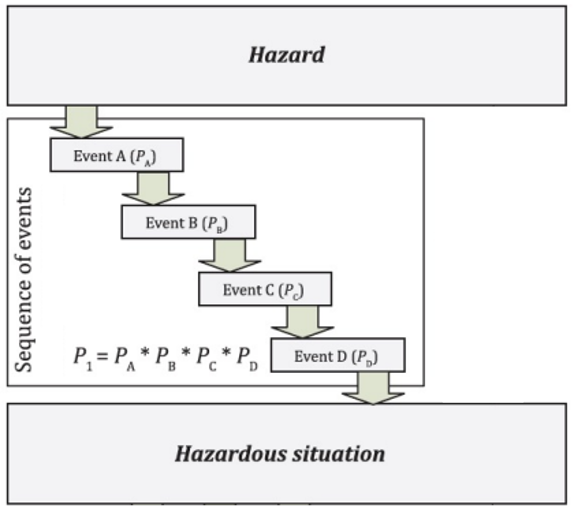
Figure 2. Relationship of hazard, sequence of events and hazardous situation, adapted from ISO/TR 24971:2020
Finally, the risks are estimated, by assigning levels of probability and severity for each hazardous situation according to the risk policy criteria. When sufficient data are available, the probability is expressed quantitatively, otherwise, a qualitative method is preferrable. Although the probability is a continuous variable, it can be decomposed in discrete levels and when it cannot be estimated, the risk is evaluated based on severity alone. The severity of a harm is a continuum, but it can also be decomposed in discrete levels. The Manufacturer decides the number of levels for both probability and severity, whereas the resulting risk matrix is often 3×3 or 5×5.
Risk evaluation
The Manufacturer evaluate each estimated risk and determine the acceptability based on the criteria defined in the risk management policy, documented in the risk management plan. In case the risk is acceptable, no further control measure will be necessary and the risk will be treated as residual.
Risk control
In case the risk is unacceptable or conditionally acceptable, it must be mitigated. There are two approaches to risk control.
The Manufacturer can approach the risk control based on the practicability of the risk measures, making trade-offs between accepting certain risks and the availability of devices on the market.
Another approach is based on the magnitude of the residual risk in which the Manufacturer tries to reduce the risk as far as possible without adversely affecting the benefit-risk ratio. There are three risk control options, which are implemented by priority order. First, the Manufacturer designs and manufacture the device inherently safe. Second, the Manufacturer adopts protective measures either on the device or in the manufacturing process. Third, the Manufacturer issues information for safety or training to users.
After implementation of risk control measures, the individual residual risks are evaluated, also using the acceptability criteria defined in the risk management policy. In case the risk remains unacceptable, and no other control measure is applicable, a benefit-risk analysis based on data and literature is performed. If the benefit does not outweigh the risk, the Manufacturer may consider modifying the device or its intended use.
Some control measures can introduce new risks or affect risks already identified, so the Manufacturer shall review the effects of risk control measures.
Evaluation of overall residual risk
After implementation and verification of risk control measures, the Manufacturer evaluates the overall residual risk, using the method and criteria defined in the risk management plan. The method to evaluate the overall residual risk can include: weighing the benefits against the overall residual risk, a visual representation of the residual risks, a comparison to similar medical devices, an expert evaluation and further investigations.
If the overall residual risk is still judged unacceptable, the Manufacturer may consider implementing additional risk control measures or modifying the device or its intended use.
Risk management review
Before releasing the device to commercial distribution, the Manufacturer confirms that the risk management has been properly executed and the results recorded as the risk management report, that the overall residual risk is considered acceptable and that appropriate methods to collect and review relevant production and post-production information are in place.
Production and post-production activities
The Manufacturer actively collects and reviews production and post-production information, including information generated during production and monitoring of production process, information generated by the user, information of installation, use and maintenance of the device, information generated by the supply chain, publicly available information and information related to the state of the art.
If the information collected is determined to be relevant to safety, the Manufacturer may apply actions concerning the medical device itself or the risk management process. The actions address devices already distributed, devices already manufactured but not distributed or devices to be manufactured.
How standards IEC 62366 correlates with ISO 14971:2019?
Section 5 of Chapter 1 of Annex I of EU MDR, requires the Manufacturer to reduce as far as possible the risks related to the ergonomic features of the device and the environment in which the device is intended to be used, and to consider the technical knowledge, experience, education, training, use environment, and the medical and physical conditions of the intended users.
Therefore, the standard IEC 62366-1:2015 assists the Manufacturer to analyse, specify, develop and evaluate the usability related to safety of a medical device. This usability engineering process allows the Manufacturer to assess and mitigate risks associated with the normal use of the device, which includes the correct use and use errors. It can also be used to identify but does not assess or mitigate risks associated with abnormal use.
The technical report IEC 62366-2:2016 has a broader focus. It focuses not only on usability related to safety, but also on usability related to attributes such as task accuracy, completeness and efficiency, and user satisfaction.
If you need more information regarding this subject, feel free to contact us at info@criticalcatalyst.com.
References:
- Regulation (EU) 2017/745, on medical devices;
- ISO 14971:2019, on application of risk management to medical devices;
- ISO/TR 24971:2020, guidance on the application of ISO 14971;
- IEC 62366-1:2015, on application of usability engineering to medical devices.











