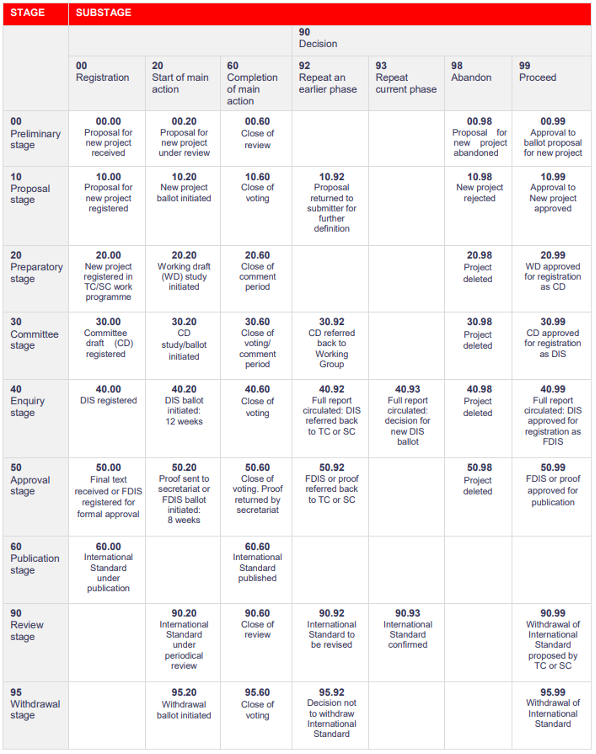
ISO Standards lifecycle
The ISO Standards lifecycle comprises 9 essential stages:
- Preliminary
- Proposal
- Preparatory
- Commitee
- Enquiry
- Approval
- Publication
- Review
- Withdrawal
Preliminary Stage (00)
This is the stage when a new idea for a Standard emerges. Firstly, it needs to be verified if that the idea has not already been suggested and recorded in the ISO/TC 211 Standards Tracker. If it has already been, then it is possible to comment or suggest additions to the existing idea.
The next step is to consider starting a preliminary work. The Technical Committee (TC) may decide by resolution or ballot, normally after recommendation from a working group, to register a Preliminary work item (PWI). The PWI is a viable outline for a future Standard, not yet sufficiently mature for further stages, and with no target dates.
A pilot study (referred as Stage 0 project) is carried out to prepare the work for further stages. The project lasts two plenary sessions, and the resulting Stage 0 report is sent to a working group. The Stage 0 report should contain an extensive outline of the proposed Standard and a draft, making it easier to continue to the next stage.
However, not all new idea comes from the internal work of the TC. A PWI can be created by national bodies, secretariat, technical management boards, advisory groups…
Proposal Stage (10)
The Proposal Stage starts with the submission of a New Work Item Proposal (NWIP) by completing the ISO Form 4. This includes defining the scope, justify why the society needs the Standard, identify the sustainable development goals, identify the stakeholders and the project leader… The detailed Standard outline and/or the initial draft must be attached to the Form 4.
ISO Standards are generally developed in parallel with CEN, under the Vienna Agreement, and if this is not appropriate, a rationale must be clearly indicated.
At the proposal stage is beneficial to identify if the project will involve Unified Modelling Language (UML) or eXtensible Markup Language (XML), once ISO has several expert groups that can help to confirm the need for expertise requirements. If there are possible complications around copyright, patents or conformity assessment they should also be raised at this stage.
To improve the draft NWIP before the formal vote, the proposal goes through an internal 30-day review by the ISO/TC 211 (Committee concerned with the standardization in the field of digital geographic information).
Based on the results of the review, the New Proposal (NP) enters the ballot period, which has a duration of 12 weeks. The committee vote decides if the NWIP can be included in the programme of work and at what stage the project will start.
Preparatory Stage (20)
The Preparatory Stage is the core of the Standard production process. The project team gets together and by consensus, creates a working draft (WD).
If there were any comments from the NP ballot, it must be demonstrated how they have been addressed. ISO/TC 211 usually use “Accepted”, “Accepted in principle” and “Not accepted”. It is necessary to elaborate on the “Accepted in principle” and “Not accepted” answers to minimize the chances for a “No” vote at a later stage ballot.
The document needs to be formatted using a specific template. Even though it is not necessary to supply the figures before the Enquiry Stage, it is a good practice to start collecting the graphical files at this point.
The project leader and the working group convenor execute a final quality assurance, and the document is ready for the Committee Stage. It is vital not to rush this submission, once the document must be almost completed for the next stage.
Committee Stage (30)
At this stage a committee draft must be ready (CD).
A CD is a document with a proper format (images etc) and with all the comments from the NP ballot resolved.
At this point it must be assured that the Terminology, UML model and XML implementation, (if applicable) are reviewed.
During the 8 weeks of consultation period, the participant members and liaisons of the TC can comment the document submitted. If there are major comments, the document can be held in the Committee Stage, corrected and submitted for a second consultation.
After the CD document is modified with the input from the consultation, a meeting with an Editing Committee (EC) is arranged. If the EC meeting reaches a consensus on how to move forward, the document can be finalized and is ready to the Enquiry Stage.
Enquiry Stage (40)
At this stage the document is called Draft International Standard (DIS).
The DIS is submitted to ISO Central Secretariat and it is then circulated not only the entire TC but also to all ISO members, with some members open the draft for public comment.
This DIS ballot period is 12 weeks long but is preceded by 8-week translation period.
The DIS is approved if two-thirds of the participant members are in favour and not more than one-quarter of the total number of votes are negative.
If there are comments, they must be addressed. Depending on the sort of changes made to resolve the comments, there are different ways to proceed:
- If there are technical changes to be made, the document must pass to an Approval Stage
- If there are no technical changes, the document proceeds to publication
- If it’s unclear whether a change is technical, the Committee Manager can help.
The document is presented again to an Editing Committee (EC) that reaches a consensus on how to move forward.
Approval Stage (50)
This stage is only applicable if a technical change is made due to Enquiry Stage comments, and it results in a Final Draft International Standard (FDIS).
If this stage is needed, the FDIS is submitted to ISO Central Secretariat. The FDIS circulates to all ISO members for an 8-week vote.
The FDIS is approved if two-thirds of the participant members are in favour and not more than one-quarter of the total number of votes are negative.
The FDIS ballot may also generate comments, however, they can only be editorial.
Publication Stage (60)
At this stage the ISO secretary submits the final document for publication.
If editorial changes were made after the FDIS ballot, a Proof is put together of how the Standard will look like upon publication. The Project leader, Convenor and Secretariat have two weeks to review the proof and raise any minor adjustments.
Review (90)
ISO Documents are all subject to systematic review, with time periods varying according to the type of document.
International Standards are reviewed every 5 years, Technical Specifications every 3 years and for Technical Reports there is no specified time.
The review is executed during a 20-week period and all ISO members are invited to respond, with ISO/TC 211 being obliged to. The vote has three main options: “Confirm”, “Amend / revise”, or “withdraw” and the decision is often made by a simple majority.
Independent of the Systematic Review any of the published Standards can be revised at any time, initiated by a resolution in the TC.
Withdrawn (95)
The decision to withdraw the Standard is not automatic.
ISO starts an 8-week Withdraw Ballot where all the ISO members are asked to confirm the withdrawal of the Standard.
Each substage has their own international harmonized stage code.