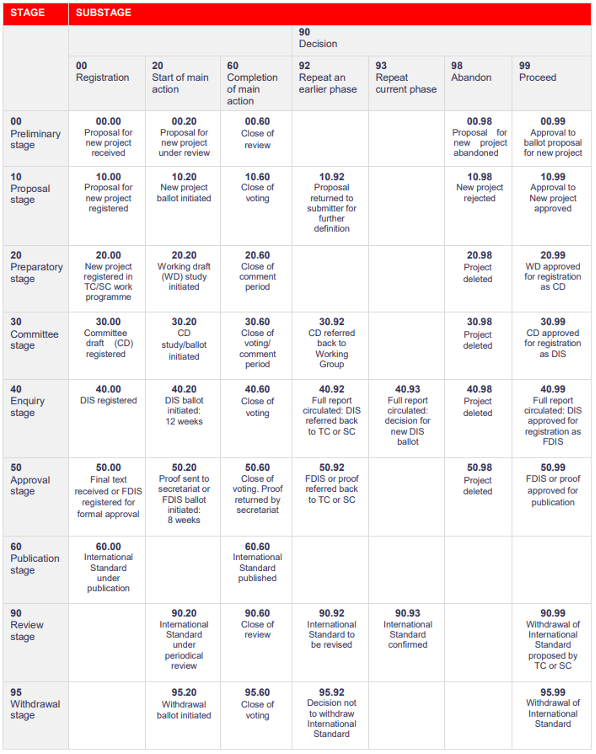
Ciclo de vida das Normas ISO
O ciclo de vida das Normas ISO inclui 9 etapas essenciais:
- Preliminar
- Proposta
- Preparatória
- Comité
- Consulta
- Aprovação
- Publicação
- Revisão
- Revogação
Etapa Preliminar (00)
É nesta etapa que surge uma nova ideia para a Norma. Primeiro, deve-se averiguar se a ideia já foi sugerida e se já encontra registada no “ISO/TC 211 Standards Tracker”. Caso já tenha sido, é possível comentar e sugerir adições à ideia existente.
O passo seguinte é começar um trabalho preliminar. A Comissão Técnica (TC) pode concluir através de uma decisão ou votação (normalmente após recomendação de um grupo de trabalho) que é necessário registar um Item de Trabalho Preliminar (PWI). O PWI é um resumo viável de uma futura Norma, sem prazos delineados e que não está suficientemente consolidado para as etapas seguintes.
Para preparar o PWI para as etapas seguintes, é realizado um estudo piloto (Stage 0 project). O projeto tem a duração de duas sessões plenárias e o relatório é enviado para um grupo de trabalho. Este relatório deve conter o resumo viável da Norma proposta e um esboço, de forma a facilitar as etapas posteriores.
No entanto, nem todas as novas ideias têm origem na TC. Um PWI pode ser criado por entidades nacionais, pelo secretariado, conselhos de gestão técnica, grupos de consulta…
Etapa da Proposta (10)
A Etapa da Proposta começa com a submissão de uma Proposta para um Novo Item de Trabalho (NWIP), preenchendo o Formulário 4 (ISO Form 4). Para isso é necessário definir o âmbito, justificar a pertinência da Norma para a sociedade, identificar os objetivos de desenvolvimento sustentável, as partes interessadas, o responsável pelo projeto… O resumo detalhado e o esboço inicial da Norma devem ser anexados ao formulário.
Normalmente as Normas ISO são desenvolvidas em paralelo com a CEN, no âmbito do acordo de Viena. Caso isso não seja apropriado, deve ser apresentado um racional.
Na Etapa da Proposta é útil identificar se o projeto envolve algum tipo de Unified Modelling Language (UML) ou eXtensible Markup Language (XML), uma vez que a ISO tem inúmeros grupos de peritos que podem confirmar a necessidade de requisitos específicos. Caso existam possíveis complicações de direitos de autor, patentes ou avaliações da conformidade, também devem ser considerados nesta etapa.
Para melhorar o esboço do NWIP antes da votação formal, a proposta passa por uma revisão interna durante 30 dias pelo ISO/TC 211 (Comissão para a harmonização no campo da informação geográfica digital).
Com base nos resultados da revisão, a Nova Proposta (NP) entra em período de votação, que tem duração de 12 semanas. A Comissão vota se o NWIP deve ser incluído no programa de trabalho e em que fase do projeto se iniciará.
Etapa Preparatória (20)
A Etapa Preparatória é fundamental para o processo de elaboração de uma Norma. Nesta etapa a equipa reúne-se e por consenso é criado um esboço de trabalho (WD).
Caso tenham existido comentários decorrentes da votação da NP, deve ser demonstrado como foram abordados. A ISO/TC 211 utiliza normalmente “Accepted”, “Accepted in principle” e “Not accepted”. É necessário elaborar os comentários que incluem “Accepted in principle” e “Not accepted” para minimizar a probabilidade de um voto contra nas votações posteriores.
O documento necessita de ser formatado utilizando um modelo específico. Apesar de não ser necessário apresentar figuras antes da Etapa de Consulta, é uma boa prática recolher referências gráficas neste ponto.
O responsável pelo projeto e o coordenador do grupo executam uma garantia da qualidade final, e o documento encontra-se preparado para a Etapa do Comité. É importante não apressar a sua submissão, uma vez que o documento deve estar quase completo para a etapa seguinte.
Etapa do Comité (30)
Nesta etapa deve estar pronto um esboço para o Comité (CD).
O CD é um documento já num formato adequado (imagens etc.) e com todos os comentários da NP resolvidos.
Nesta altura deve ser assegurada a revisão da Terminologia, modelo UML e implementação XML (se aplicável).
Durante as 8 semanas do período de consulta, os membros participantes e os elementos de ligação do TC podem comentar o documento submetido. Caso existam comentários major, o documento pode ser mantido na Etapa do Comité, corrigido e submetido uma segunda vez.
Após o documento ser modificado, é realizada uma reunião com a Comissão de Edição (EC). Caso a EC chegue a um consenso quanto ao seguimento do projeto, o documento pode ser finalizado e fica pronto para a Etapa de Consulta.
Etapa de Consulta (40)
Nesta fase o documento designa-se por “Esboço de Norma Internacional” (DIS).
O DIS é submetido ao Secretariado Central da ISO, que o faz circular não apenas entre os membros do TC, mas entre todos os membros ISO, que o podem inclusive disponibilizar para consulta pública.
O período de votação tem a duração de 12 semanas, precedidas por 8 semanas dedicadas à tradução do documento.
O DIS é aprovado caso dois terços dos membros participantes votem a favor e caso não mais do que um quarto do número total de votos seja negativo.
Caso existam comentários, estes devem ser respondidos. Existem formas diferentes de proceder, dependendo das alterações realizadas para resolver os comentários:
- Caso tenham sido realizadas alterações técnicas, o documento passa por uma Etapa de Aprovação;
- Caso não tenham sido realizadas alterações técnicas, o documento passa diretamente para Publicação;
- Caso não seja claro se as alterações são técnicas, a decisão é auxiliada pela Comissão de Gestão.
O documento é apresentado novamente à Comissão de Edição, que chega a um consenso quanto ao seguimento do projeto.
Etapa de Aprovação (50)
Esta etapa é aplicável apenas caso tenha sido efetuada uma alteração técnica no decorrer da Etapa de Consulta. Nesta etapa é elaborado um “Esboço Final da Norma Internacional” (FDIS).
Caso esta etapa seja necessária, o FDIS é submetido ao Secretariado Central da ISO, que o faz circular entre todos os membros ISO para um período de votação de 8 semanas.
O DIS é aprovado caso dois terços dos membros participantes votem a favor e caso não mais do que um quarto do número total de votos seja negativo.
A votação do FDIS também pode gerar comentários, mas têm que ser meramente sobre aspetos relacionados com a sua redação.
Etapa de Publicação (60)
Nesta etapa, a secretaria da ISO submete o documento final para publicação.
Caso tenham sido efetuadas alterações quanto à redação do FDIS, é elaborado uma prova para demonstrar o aspeto do documento final após publicação. O responsável pelo projeto, o coordenador e o secretariado dispõem de 2 semanas para rever a prova e realizar pequenos ajustes.
Revisão (90)
Os documentos ISO são sujeitos a uma revisão sistemática, que pode variar dependendo do tipo de documento.
As Normas Internacionais são revistas a cada 5 anos, as Especificações Técnicas a cada 3 anos e os Relatórios Técnicos não têm um prazo específico para revisão.
A revisão é executada ao longo de um período de 20 semanas e todos os membros ISO são convidados a participar, exceto os membros da ISO/TC 211, que são obrigados. O voto inclui três opções principais: “Confirmar”, “Alterar/ rever” ou “Revogar”, e a decisão final é obtida por maioria simples.
Independentemente do processo de revisão sistemática, qualquer Norma publicada pode ser revista a qualquer altura, mediante decisão da TC.
Revogação (95)
A decisão de revogar uma Norma não é automática.
A ISO começa uma votação de 8 semanas, na qual é solicitado aos membros para confirmarem a revogação da Norma.
Cada sub-etapa tem um código internacional harmonizado específico.