


EUDAMED
A EUDAMED permitirá uma melhor caracterização do ciclo de vida dos dispositivos médicos disponíveis no mercado da União Europeia (UE), aumentando a transparência através de um melhor acesso à informação e facilitando a coordenação entre os Estados Membro da UE.
A EUDAMED será constituída por seis módulos:
- Registo de operadores económicos (fabricantes, produtores de sistemas e conjuntos, mandatários e importadores);
- Registo de dispositivos/Base de dados UDI;
- Organismos notificados e certificados;
- Investigações clínicas;
- Vigilância e monitorização pós-comercialização;
- Fiscalização do mercado.
Em 2019, a Comissão Europeia publicou uma nota esclarecendo que a plataforma EUDAMED só estará completamente operacional quando todos os módulos estiverem disponíveis e de acordo com as especificações funcionais, confirmadas num relatório de auditoria independente.
O lançamento da plataforma EUDAMED estava planeado para 2020, mas a data prevista sofreu alguns adiamentos.
Novo cronograma da EUDAMED
Em junho de 2022, a Comissão Europeia publicou o planeamento do novo cronograma para implementação da EUDAMED.
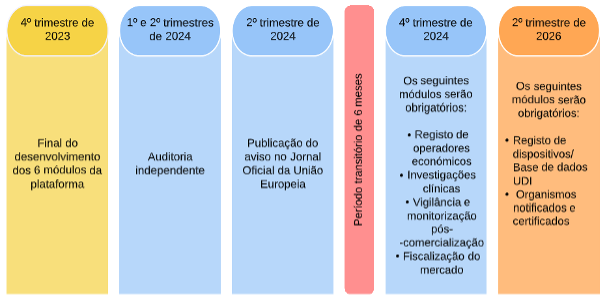
Figura 1. Novo cronograma da EUDAMED
(adaptado de “EUDAMED Timeline – The European Commission planning – June 2022”)
É expectável que uma versão completa minimamente viável da EUDAMED seja desenvolvida até ao último trimestre de 2023.
A auditoria independente realizar-se-á nos dois primeiros trimestres de 2024.
Espera-se que o aviso de completa operacionalização da EUDAMED seja publicado no Jornal Oficial da União Europeia no segundo trimestre de 2024.
Após a publicação do aviso, decorrerá um período transitório de 6 meses, com término no último trimestre de 2024. A partir desse momento, é obrigatório cumprir com os requisitos relativos aos módulos de registo de operadores económicos, investigações clínicas, vigilância e monitorização pós-comercialização e fiscalização do mercado. Todos os módulos da EUDAMED serão obrigatórios no segundo trimestre de 2026.
Referências:
- EUDAMED Timeline – The European Commission planning – June 2022.











