


O que é um produto de combinação de medicamento-dispositivo?
Produtos de combinação de medicamentos-dispositivos (DDCs) são medicamentos que contêm um ou mais dispositivos médicos como parte integral da sua composição ou produtos que não incorporam dispositivos de forma integral, mas que necessitam de dispositivos médicos para a sua administração, doseamento ou utilização.
Definições importantes
A combinação de um medicamento com um dispositivo médico pode constituir um produto integral ou um produto não-integral.
Produtos integrais – Um produto integral pode ser:
- Um dispositivo que incorpore uma substância como parte integral, e que essa substância, se utilizada separadamente, seja considerada um medicamento e seja responsável pela ação principal do produto. Exemplos incluem medicamentos com sensores integrados.
- Um dispositivo no qual a finalidade prevista seja administrar um medicamento e que constitua um produto individual integral destinado exclusivamente à utilização em combinação e que não seja reutilizável. Exemplos incluem seringas de utilização única pré-cheias, dispositivos intrauterinos de libertação de medicamentos e inaladores de utilização única pré-cheios.
Produtos não-integrais – Os componentes (medicamento e dispositivo) de produtos não-integrais não são fisicamente incorporados durante o fabrico, mas são combinados para administração. Os produtos não-integrais incluem produtos embalados conjuntamente e produtos referenciados:
- Embalados conjuntamente – O medicamento e o dispositivo médico são incluídos no mesmo pack e são colocados no mercado conjuntamente.
- Referenciados – As informações do medicamento (RCM e/ou folheto informativo) fazem referência ao dispositivo médico que deve ser utilizado, mas este é obtido separadamente.
Exemplos de produtos não-integrais incluem: copos, colheres e seringas para administração oral, injetores e canetas recarregáveis por cartuchos, nebulizadores, vaporizadores, bombas para administração de medicamentos e dispensadores eletrónicos de medicamentos.
Quadro regulamentar
Os produtos integrais são regulados pela legislação farmacêutica da UE (Diretiva 2001/83/EC ou Regulamento (CE) N.º 726/2004), ao abrigo da qual, o medicamento deve obter uma autorização de introdução no mercado. O Artigo n.º 117 do RDM indica que o dossiê de autorização de introdução no mercado do medicamento deve incluir evidência de conformidade da parte do dispositivo com os Requisitos Gerais de Segurança e Desempenho (RGSDs) (Anexo I do RDM). O pedido de autorização de introdução no mercado deve incluir o certificado CE, ou, caso o dispositivo ainda não tenha marcação CE e seja necessária a intervenção de um organismo notificado de acordo com o RDM, o requerente deve incluir um parecer emitido pelo organismo notificado sobre a conformidade do dispositivo com os RGSDs.
No entanto, caso o dispositivo incorpore uma substância que, se utilizada separadamente, seja considerada um medicamento, mas a ação dessa substância seja auxiliar à ação do dispositivo, o produto deve ser avaliado e autorizado de acordo com o RDM. Para determinar se a substância é “auxiliar” é considerada a disponibilidade da substância no corpo humano e/ou a quantidade da substância disponível.
Caso os produtos sejam embalados conjuntamente ou referenciados, o medicamento é regulado pela Diretiva 2001/83/EC ou pelo Regulamento (CE) N.º 726/2004, e o dispositivo deve obter marcação CE de acordo com o RDM.
Se o dispositivo tiver a finalidade de administrar um medicamento, o produto é regulado pelo RDM. Ainda assim, o medicamento deve cumprir com as disposições da Diretiva 2001/83/EC ou do Regulamento (CE) N.º 726/2004. No entanto, se o dispositivo tiver a finalidade de administrar um medicamento, mas constitua um produto individual integral para uso exclusivo em combinação e não reutilizável, o produto é regulado pela Diretiva 2001/83/EC e pelo Regulamento (CE) N.º 726/2004, e a parte relativa ao dispositivo deve incluir evidência de conformidade com os RGSDs aplicáveis.
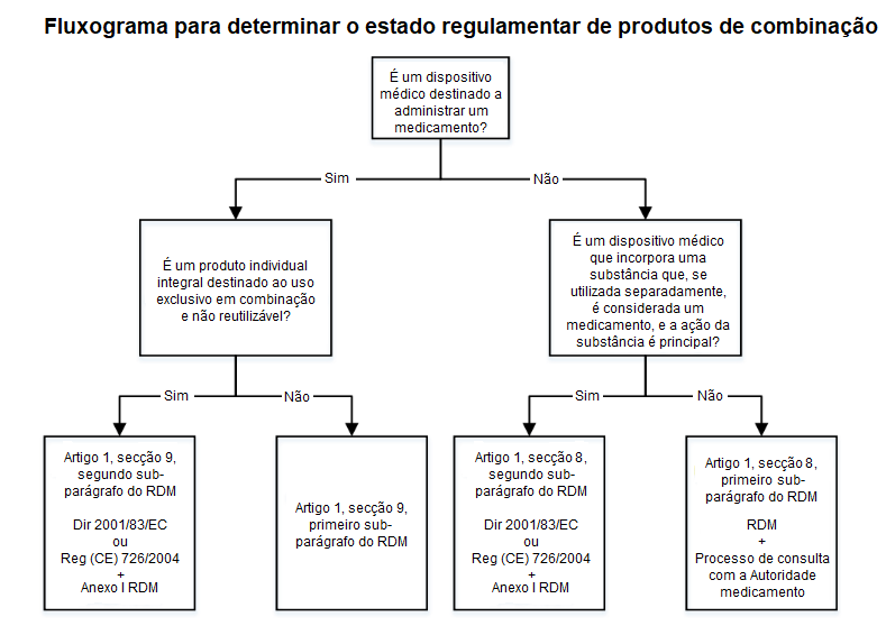
Adaptado de: MDCG 2022–5 Orientação sobre a fronteira entre dispositivos médicos e medicamentos sob o Regulamento (UE) 2017/745 relativo aos dispositivos médicos, Abril 2022.
Desafios
O desenvolvimento de produtos combinados requer uma abordagem colaborativa. Geralmente, os promotores têm conhecimentos no mercado farmacêutico ou no mercado dos dispositivos médicos, o que pode ser um desafio, uma vez que necessitam de adquirir expertise antes de começar a desenvolver os seus produtos.
Na decisão sobre o quadro regulamentar aplicável a uma combinação de produtos, o primeiro passo é estabelecer se o produto é integral. O passo seguinte na abordagem à estratégia regulamentar é definir claramente a finalidade prevista e o modo de ação do produto, percebendo a parte da combinação que é principal e a parte que é auxiliar.
As DDCs são associadas a um aumento do consumo de tempo e de recursos, uma vez que é necessário assegurar que o desempenho e a segurança são alcançados para todos os componentes do produto, de forma individual e em conjunto.
Referências:
- Regulamento (UE) 2017/745, relativo aos dispositivos médicos
- Agência Europeia do Medicamento (EMA) – Orientação sobre a documentação relativa à qualidade de medicamentos quando utilizados com um dispositivo médico, 22 Julho 2021
- MDCG 2022 – 5 Orientação sobre a fronteira entre dispositivos médicos e medicamentos sob o Regulamento (UE) 2017/745 relativo aos dispositivos médicos, Abril 2022











