


What is a drug-device combination product?
Drug-device combination products (DDCs) are medicinal products which contain one or more medical devices as an integral part of their composition or medicinal products which do not incorporate medical devices in an integral manner, but which require medical devices for their administration, correct dosing or use.
Relevant definitions
The combination of a medicinal product and a medical device can constitute an integral product or a non-integral product.
Integral products – An integral product can be:
- A device which incorporates a substance as an integral part that, if used separately, is considered a medicinal product and the substance action is principal. Examples include medicinal products with embedded sensors.
- A device intended to administer a medicinal product that constitutes a single integral product intended exclusively for use in combination and which is not reusable. Examples include single-use pre-filled syringes, drug-releasing intra-uterine devices and single-use pre-filled dry powder inhalers.
Non-integral products – The components (medicinal product and device) of non-integral products are not physically integrated during manufacturing, but are combined for administration. Non-integral products include co-packaged products and referenced products:
- Co-packaged – The medicinal product and the medical device are placed on the market packed together into a single pack.
- Referenced – The medicinal product information (SmPC and/or package leaflet) refers to a specific medical device to be used which is obtained separately.
Examples of non-integral DDCs include: cups, spoons and syringes for oral administration, refillable pens and injectors using cartridges, nebulisers, vaporisers, pumps for medicinal product delivery and electronic tablet dispensers.
Regulatory framework
Integral products are regulated by EU pharmaceutical legislation (Directive 2001/83/EC or Regulation (EC) No 726/2004), under which the medicinal product must obtain a marketing authorisation. Regarding the device part, Article 117 of the MDR states that it should be included evidence of the conformity with the relevant General Safety and Performance Requirements (GSPRs) (MDR Annex I). With this Article, marketing authorisation application should include a CE certificate for the device or, in case it is not CE marked and the involvement of a notified body is required according with the MDR, the applicant must include an opinion from a notified body on conformity of device with the GSPRs.
However, if the device incorporates a substance as an integral part that, if used separately, is considered a medicinal product, but the action of the substance is ancillary to that of the device, the product shall be assessed in accordance with the MDR requirements. Determining whether the substance “is ancillary” shall consider the substance availability in the human body and/or the quantity that is available.
If the products are co-packaged or referenced, the medicine is regulated by Directive 2001/83/EC or Regulation (EC) No 726/2004, and the device part must be CE marked in accordance with the MDR.
In case the device is intended to administer a medicinal product, it is regulated by the MDR, but the medicinal product part is still governed by Directive 2001/83/EC or Regulation (EC) No 726/2004. However, if the device is intended to administer a medicinal product, but it constitutes a single integral product intended exclusively for use in combination and not reusable, the product is regulated by Directive 2001/83/EC and Regulation (EC) No 726/2004, and the device part must include evidence of the conformity with the relevant GPSRs.
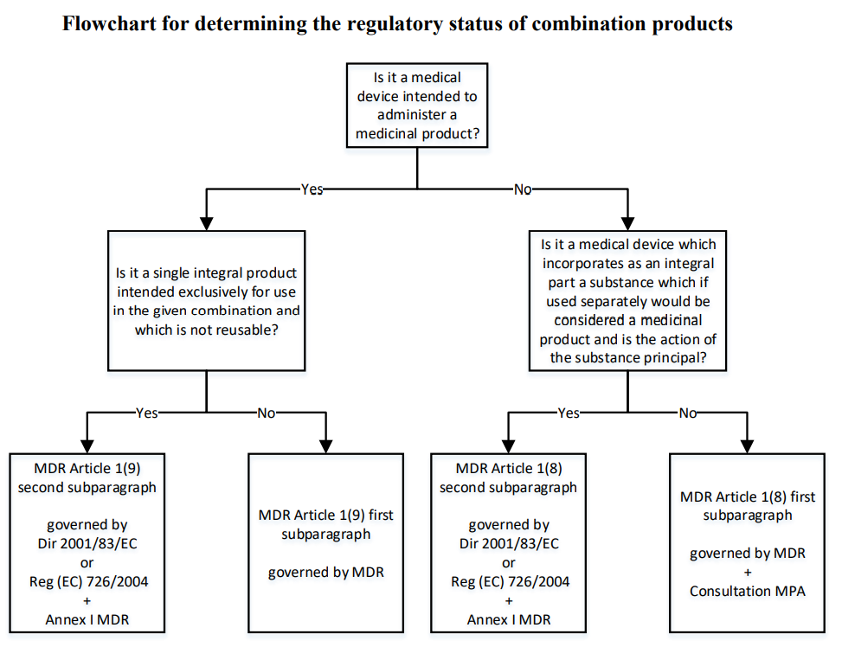
Source: MDCG 2022–5 Guidance on borderline between medical devices and medicinal products under Regulation (EU) 2017/745 on medical devices, April 2022
Challenges
The development of combination products requires a collaborative approach. Sponsors have typically expertise in pharmaceutical market or in medical devices market, and that can pose a challenge once they have to acquire the necessary expertise prior to start developing their products.
When deciding on the regulatory status of a combination product, the first step is to establish whether the product is an integral product. Other essential step in the regulatory approach to combination products is to clearly define the product intended purpose and mode of action to understand which part of the combination is principal and which is ancillary.
DDCs can also be associated with higher resources consumption and time expenditure, because it is necessary to assure that performance and safety are met for both parts of the product, individually and in combination.
References:
- Regulation (EU) 2017/745, on medical devices
- European Medicines Agency (EMA) Guideline on quality documentation for medicinal products when used with a medical device, 22 July 2021
- MDCG 2022 – 5 Guidance on borderline between medical devices and medicinal products under Regulation (EU) 2017/745 on medical devices, April 2022











