


The Covid-19 pandemic, the shortages of raw materials caused by the Russian invasion of Ukraine and the low notified body capacity has put a strain on market readiness. It is expected the number of MDR certificates to be approximately 7,000 by May 2024, which is significatively lower than the 22,793 valid certificates issued under the Directives that will expire in that date.
Thus, at the Employment, Social Policy, Health and Consumer Affairs (EPSCO) Council back on June 2022, some Health Ministers expressed their concerns that the implementation of the MDR could threaten the availability of certain medical devices and jeopardise the access of innovative technologies to the EU market. The Ministers called on the Medical Device Coordination Group (MDCG) to propose solutions for the insufficient capacity of notified bodies and the low level of preparedness of manufacturers. The MDCG published the document MDCG 2022-14 on August 2022, laying out 19 actions to enhancing notified body capacity, access to notified bodies and manufacturers’ preparedness.
Despite their full support for the MDCG position paper 2022-14, several Member States, some Members of the European Parliament and stakeholders consider that those actions will not be sufficient and are calling for additional measures, namely an urgent, targeted legislative initiative to amend the MDR.
The European Commission has listened to the concerns and showed ready to act, presenting a proposal with solutions to the EPSCO Council on 9 December 2022.
The presented elements of a legislative proposal for an amendment of the MDR and IVDR include:
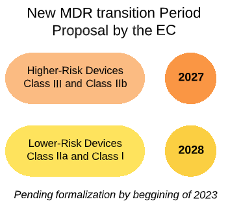
- An extension of the transitional period [MDR Article 120(3)] with deadlines depending on the risk class of the device. Those deadlines could be 2027 for high-risk devices (class III and class IIb) and 2028 for low-risk devices (class IIa and class I);
- The extension of the transitional period could be combined with an extension of the validity of certificates issued under the Directives;
- Conditions to ensure that the extension applies only to devices that do not present unacceptable risks to health and safety, and that not undergone significant changes in design or intended purpose, and for which the manufacturers have already undertaken the necessary steps to launch the certification process under the MDR. These steps include an adaptation of their quality management system to the MDR and submission and/or acceptance of the manufacturer’s application for conformity assessment by a notified body before a certain deadline (e.g. 26 May 2024);
- The removal of the ‘sell off’ provision [MDR Article 120(4) and IVDR Article 110(4)].
The specific legislative changes have not yet been announced. The extension of validity of certificates issued under the Directives, and if the proposed changes will affect in vitro diagnostic medical devices is still unclear.
The Commission will update the text and provide an MDCG position paper on the expiring of certificates. The legislative process to change the text is expected to begin in early 2023.
The topics for EPSCO Council on 9 December 2022 can be found here!
The recording of the meeting can be found here!











